![[Translate to Englisch:] eine Hand unterschreibt ein regulatorisches Dokument, auf dem eine rote Brille liegt](/fileadmin/user_upload/content-bilder/Branchenl%C3%B6sungen/Pharma/pharma.regulations/Regularien_800x500_-_Kopfbild.jpg)
The quality requirements and checks for medical devices and pharmaceutical products and their production processes are stricter than in any other industry. Manufacturers have an obligation to guarantee both product quality and the integrity of the test data obtained by an automated inspection.
To do this, many complex requirements from national and international standards, laws and directives must be adhered to. Machine vision based inspection systems for quality assurance fall into the “computerised systems” category and are therefore subject to particularly exacting requirements.
In the pharmaceutical industry, terms such as cGMP, GAMP, audit trails and CFR conformity are widely known in this context. The information below explains what regulations apply to automated quality assurance. As a certified expert in inspection systems for the pharmaceuticals and healthcare industries, OCTUM has the relevant expertise and experience to offer you competent support in the creation of your quality assurance inspection systems. Why not contact us today!
Any questions?
Get directly in touch with our
experts on +49 7062 91494-0
or write to us using our contact form
Automated in-process quality assurance is subject to regulations on three levels. We are familiar with these requirements and offer you peace of mind with maximum quality.
Here, you’ll find an overview of the various applicable regulations and how they fit together: from laws and acts passed by individual countries to directives and also recommendations. With OCTUM, you can trust in a reliable and experienced partner for inline quality assurance with vision inspection systems.
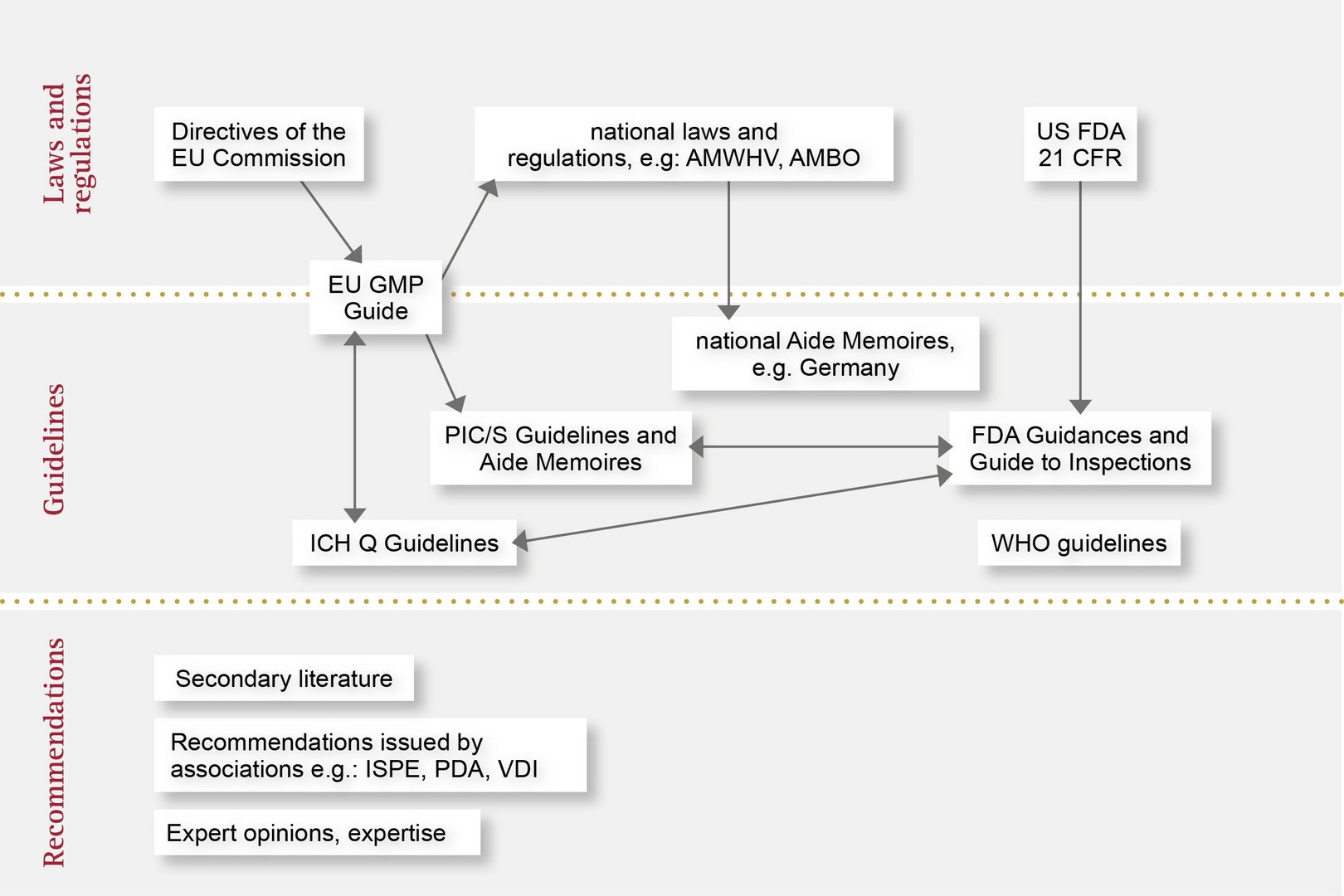
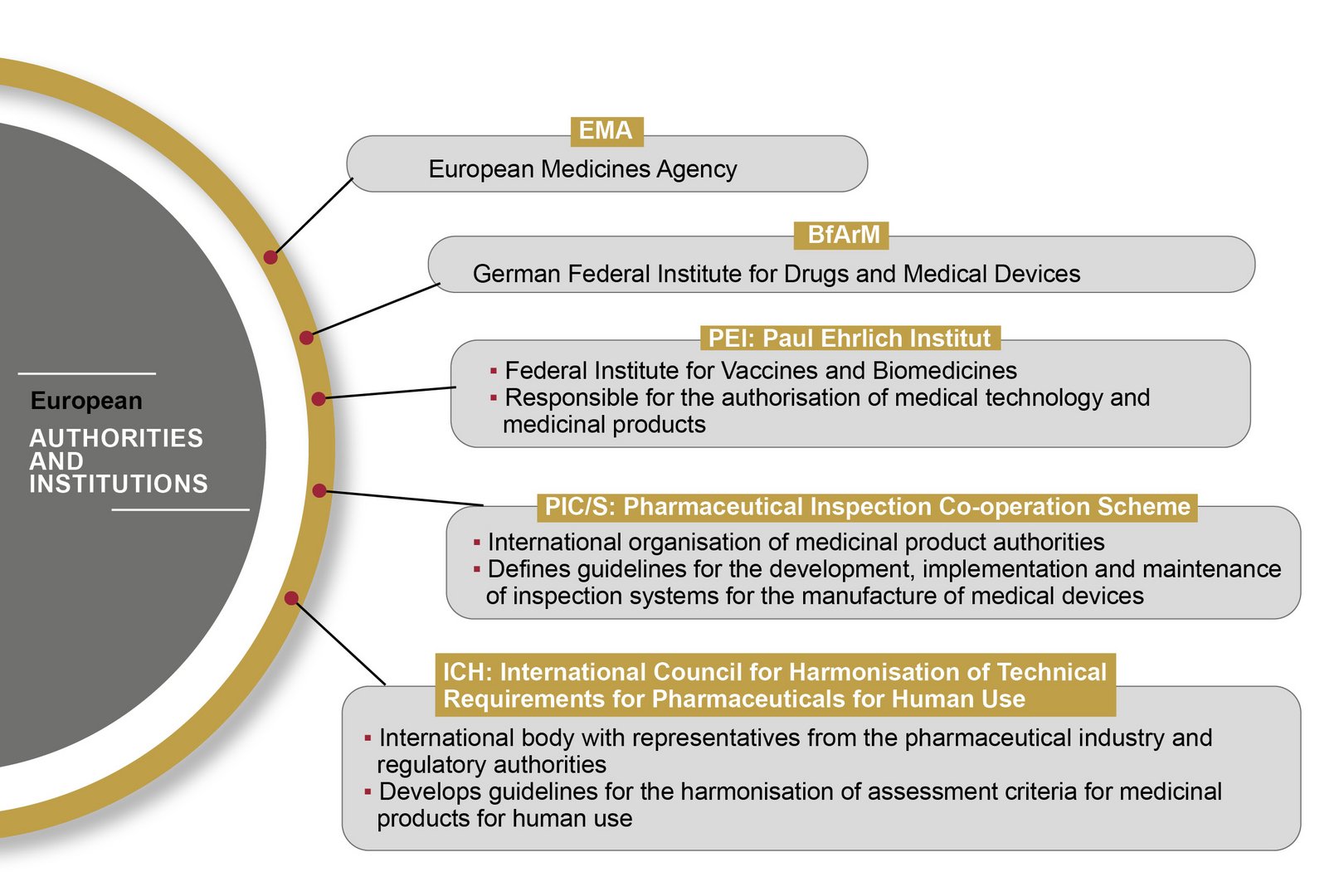
In the European Union, the handling of medical devices and pharmaceutical products is standardised and regulated by the European Medicines Agency (EMA). The EMA publishes guidelines based on EU Commission directives, which are transposed into national law by the medical regulatory bodies of the individual Member States.
A great many regulations and recommendations must be observed during the design and manufacture of automated inspection systems. For example, similarly to CFR 21 Part 11, the EU GMP Annex 11 guideline describes the requirements for computerised systems, but also extends to cover the topics of qualification and validation. The latter is set out in detail in the associated Annex 15 guideline.
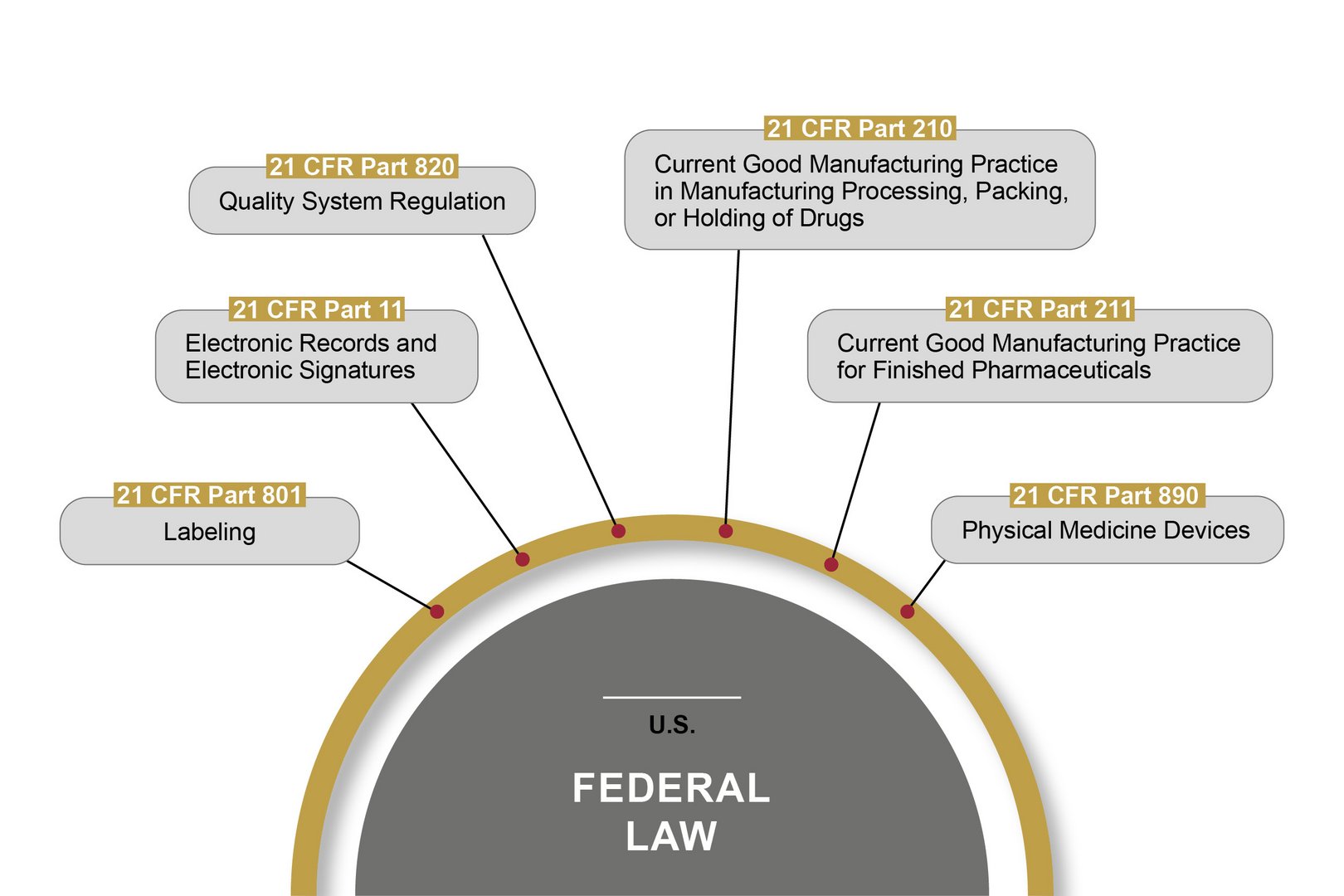
The FDA (Food and Drug Administration) is the U.S. authority responsible for the central marketing authorisation and monitoring of pharmaceutical products and medical devices. The FDA monitors the market more proactively than European authorities – with on-site inspections, for example. Therefore, OCTUM solutions are always based on the most stringent rules and regulations.
21 CFR Part 11 is the best known American federal law passed by the FDA in this field. It lays down the requirements for computerised systems that handle pharmaceutical products, among other things. These cover the prerequisites for the reliability and trustworthiness of digital documents, in the same way as traditional records. Part 11 defines the conditions under which the electronic records and signatures of computerised systems can be considered trustworthy and reliable. Certain other parts of 21 CFR also have to be observed.
The “Good Automated Manufacturing Practice 5” guideline is regarded as the standard regulatory volume for the validation of computerised systems in pharmaceutical and medical technology. It lays down requirements for planning, commissioning, validation and qualification – of production systems, for example. The guideline is published by the International Society of Pharmaceutical Engineers (ISPE), which has committees including representatives from medical regulatory bodies and experts from the pharmaceuticals industry.
The GAMP5 is not a law, but rather a recommended methodology for developing a compliant computerised system. The GAMP5 Software Development Life Cycle describes the requirements from the planning to the productive operation of automated systems, under consideration of all the expected risks.
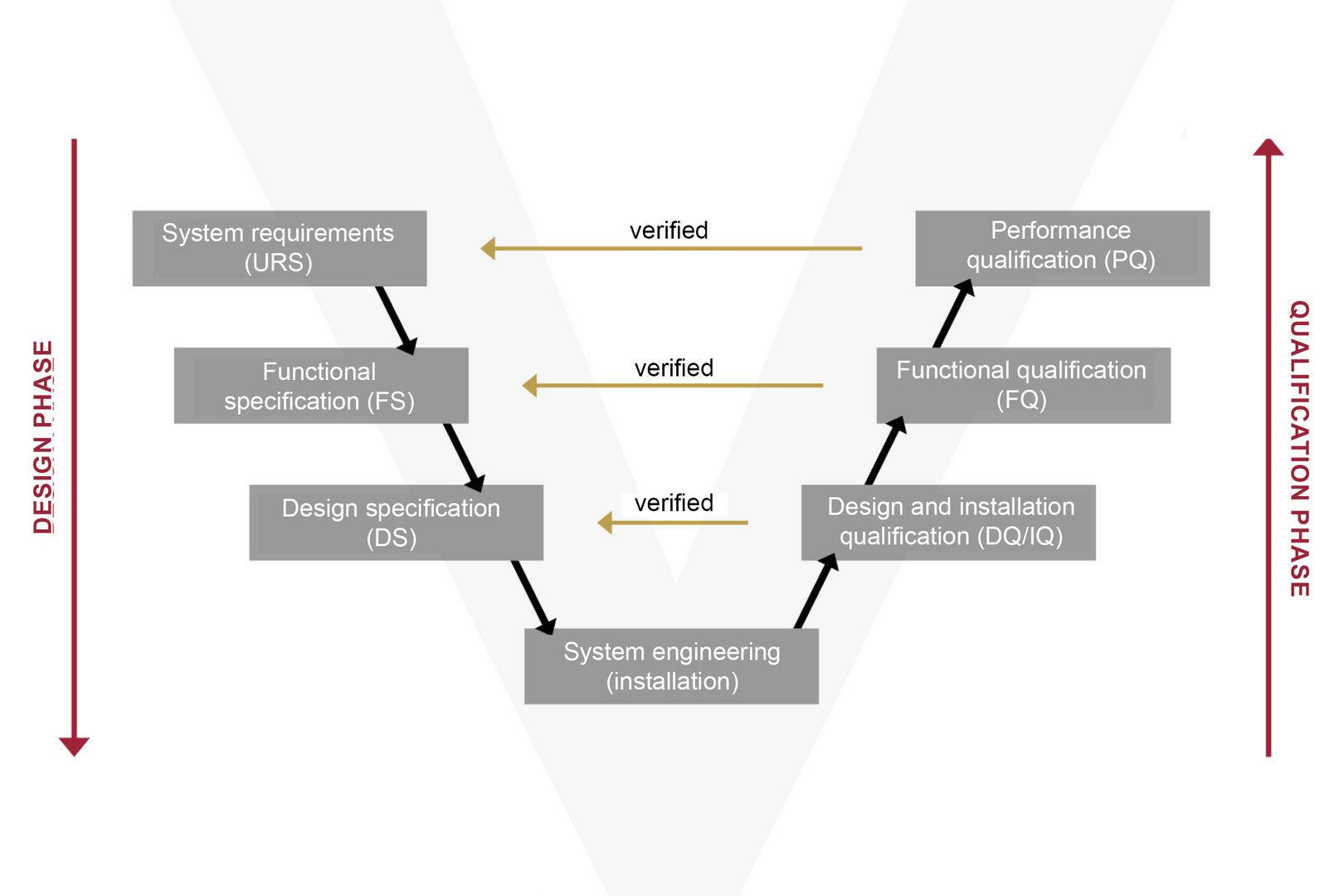
In practice, the system’s function (FS) and design (DS) are conceived with direct reference to the URS (User Requirement Specification). Following a specific risk analysis, the system is implemented and put into operation in the machine. During the subsequent qualification phase, the Installation, Operational and Performance Qualification (IQ/OQ/PQ) process verifies and records whether the overall system complies with the specifications.
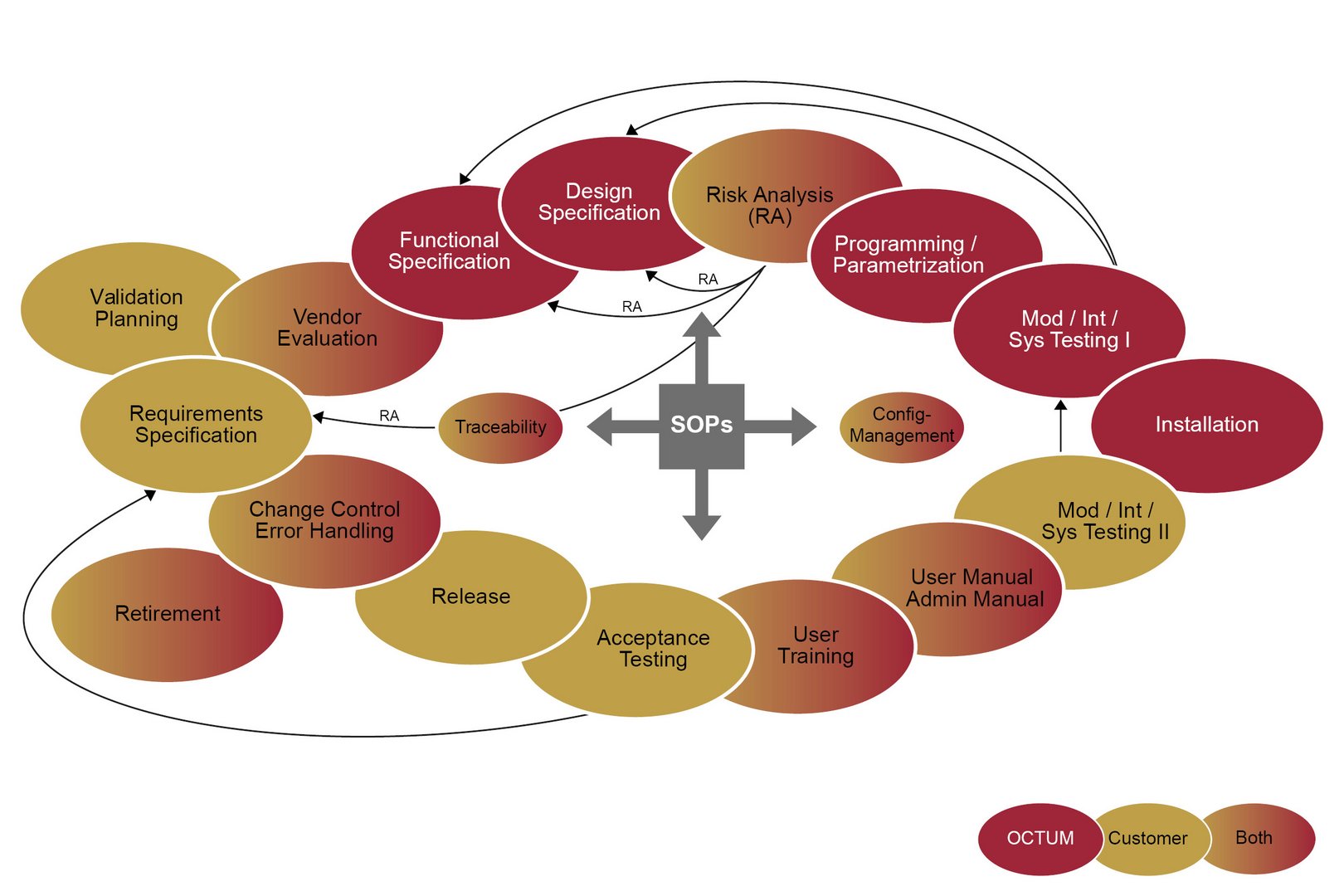
Inspection systems from OCTUM have been specially developed for the pharmaceuticals industry and have been an established part of production since 1996. Thanks to our many years of experience in the fields of vials, syringes, pipettes and bandages, among other items, our solutions are specially tailored to the exacting requirements of this industry and satisfy all the necessary regulations. Our range of services extends from consulting and planning to the implementation and qualification of the compliant overall system – including all the documents the regulations require.